
【复合材料信息】异核双原子催化剂动态调控新突破,电场控制下的“分分合合”






选题背景
动态调节催化中心结构和电子态在电催化反应中对提高催化性能具有重要意义。与传统的固定结构催化剂相比,可以响应外界刺激,实现可逆结构变化的系统为实现“按需”调整反应路径和选择性提供了可能性。然而,如何控制和重构单原子到双原子活性中心,并实时跟踪其构型演变和催化行为,仍然是当前材料设计和机制研究中的关键挑战。
本文基于1Tʹ-MoS₂聚集在这个硫位置的二维材料构建了一个高负荷的Cu和Pt单原子催化位点,通过施加外部电场可逆地重构为Cu。–双原子催化结构Pt异核(DAC)。原点同步辐射 x光吸收谱(XAS)以及密度泛函理论(DFT)以及分子动力学模拟(MD)明确揭示,S–H键的质子化过程促进金属化–硫磺键断裂和重排,从而实现DAC构型的可控生成和释放。这种动态DAC催化剂在苯乙炔的半氢化反应中表现出远远超过单原子系统的活性、选择性和稳定性。
这项工作不仅提供了电场诱导原子级结构重构的实验模式,还展示了动态活性中心在反应过程中如何协调反应路径,为电催化剂可编程设计和结构效率关系研究提供了新的理论依据。
来自新加坡国立大学化学系的罗健平教授和博士生吴建花(第一作者)首次讨论了电催化过程中异核双原子在电场作用下的动态调节。合作伙伴包括香港理工大学应用物理系的杨明教授、新加坡科技研究局(A*STAR)席识博士,化学、能源和环境可持续发展研究所。这里的“合作者”是指共同通信的作者。
科学亮点
1. MoS采用电化学拔硫技术。₂表面制造大量位置,形成“原子级锚点”,成功承载超高密度铜。(Cu)和铂(Pt)单原子,为后续动态重组奠定基础。
2. 电场控制下的原子级重组:传统催化剂一旦准备好,结构就会保持不变。然而,这项研究打破了这种限制——Cu和Pt单原子可以在电场控制下可逆结合/分离,形成高活性的异核双原子催化剂(Cu-Pt DACs)!这个团队首次实现了Cu–二维MoSPt异核双原子催化剂₂表面电场诱导结构,具有动态可逆结构转换能力;
团队发现,通过原点X射线吸收光谱和理论计算:
在MoS上施加负电压时,Cu接近Pt单原子。₂异核双原子结构表面形成稳定的表面(Cu-Pt DACs)键长仅2.6 Å;
关闭电场后,他们又“各奔东西”,恢复到独立的单原子状态。
这个过程就像一个“原子开关”,完全可逆,高度可控!
3. 协同效应:1 1 > 2
采用原点光谱 根据理论计算,团队发现双原子协同调节吸附能量。Cu负责强吸附炔烃,Pt负责高效活性氢。两者结合后,中间体结合可以恰到好处,加速反应,避免过度加氢。
图文解析
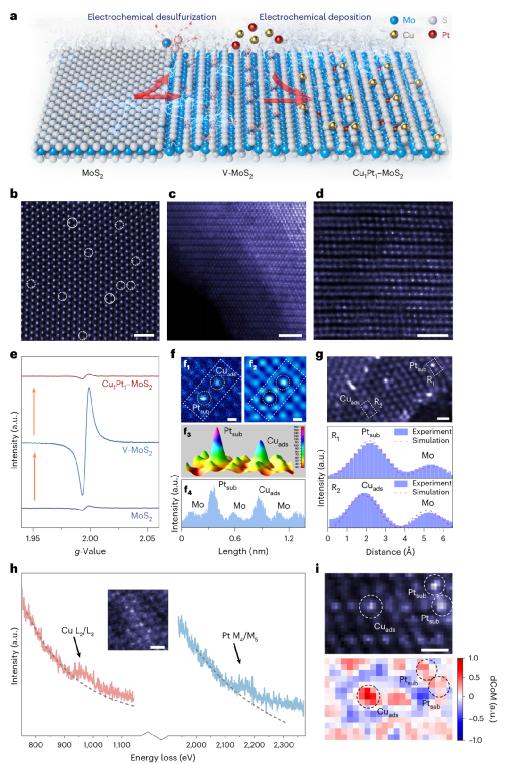
图 1. DAC 构建前驱结构和单原子导向行为的多尺度表征。
图1系统显示了异核双原子催化剂(DAC)在原子尺度下构建前驱过程和空间定位策略。MoS通过电化学脱硫在MoS中₂将高密度硫位置引入表面,诱导2H相向1Tʹ相转,显著提高其对金属单原子的导向能力。通过HAADFF形成这种变化和位置结构。–STEM显像、EPR谱和拉曼光谱等多种方法得到证实。随后选择电沉积方法,将Cu和Pt分别负载到MoS的位置。₂单原子结构表层,形成高负荷(Cu: 7.9 wt%,Pt: 6.7 wt%)。结合EELS分析,4D-STEM显示Cu原子主要位于Mo位(吸附位),Cu_ads),而且Pt原子表现出明显的替代混合行为。(Ptsub)。电荷密度投射进一步表明Cu位点具有较好的正电密度分布,这表明它更容易参与电荷转移和成键行为。上述异质位点的空间相邻性是由电场诱导的Cu–在为后续构效关系研究奠定微观前提的同时,Pt成键提供了结构基础。
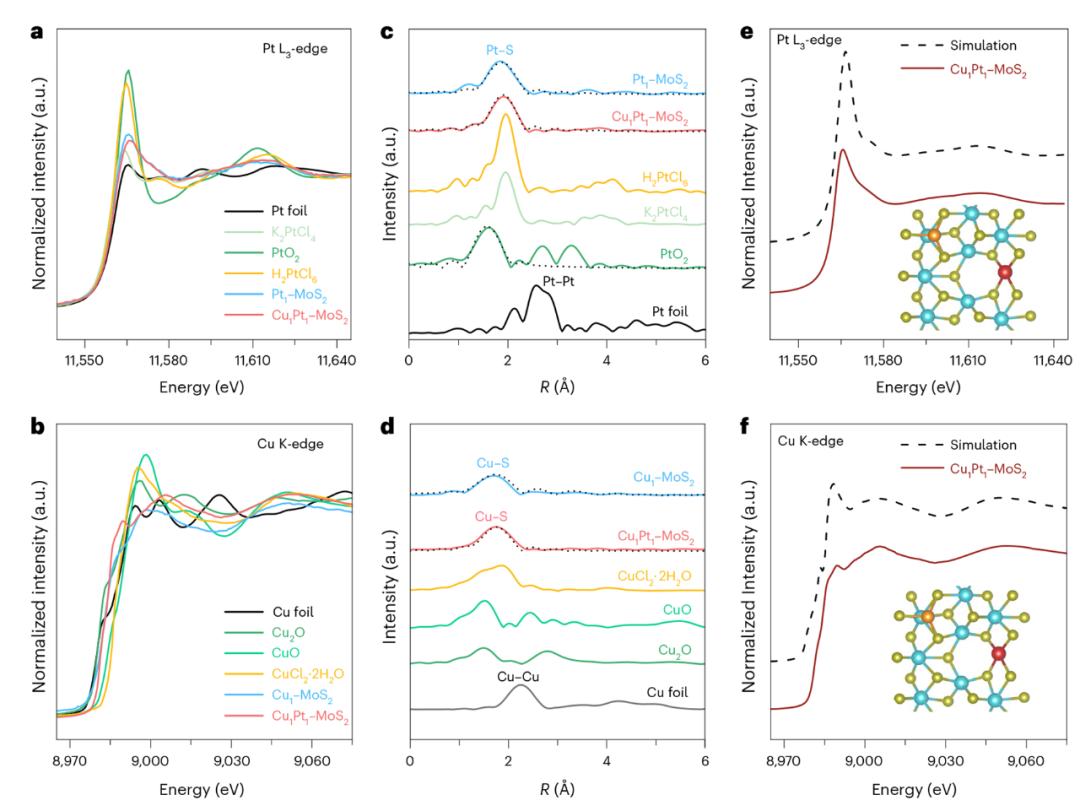
图 2. 异核前驱结构通过单原子配位环境和电子态分析得到验证。
图2通过同步辐射X射线吸收近边结构。(XANES)扩展X射线吸收精细结构。(EXAFS)MoS中Cu和Pt的谱系₂定量分析表面的配位环境及其分散态。XANES谱显示,Pt显示L₃-edge处的白线强度明显低于PtO。₂及H₂PtCl₆参考样,表明其处于较低的氧化状态( 2);并且Cu的K-edge谱证实了它是 1价态,符合其三配位吸附构型。进一步揭示了EXAFS傅立叶转换谱。–S(~1.7 Å)与Pt–S(~1.9 Å)配置距离,没有金属–金属透射信号验证了金属单原子的独立分布。通过建立Cu_,理论建模进一步支持实验发现。ads–Pt与实验XANES谱相结合的_sub模型,获得高度匹配的理论曲线。此外,模拟结果显示,Pt更容易与S形成稳定的四配位结构,而Cu则侧重于以较低的配位数吸附表面,电子结构与配位环境高度协同。对异核DAC的可能构型进行了明确的整体结果,并为电场响应过程中的动态调控提供了准确的结构参考。
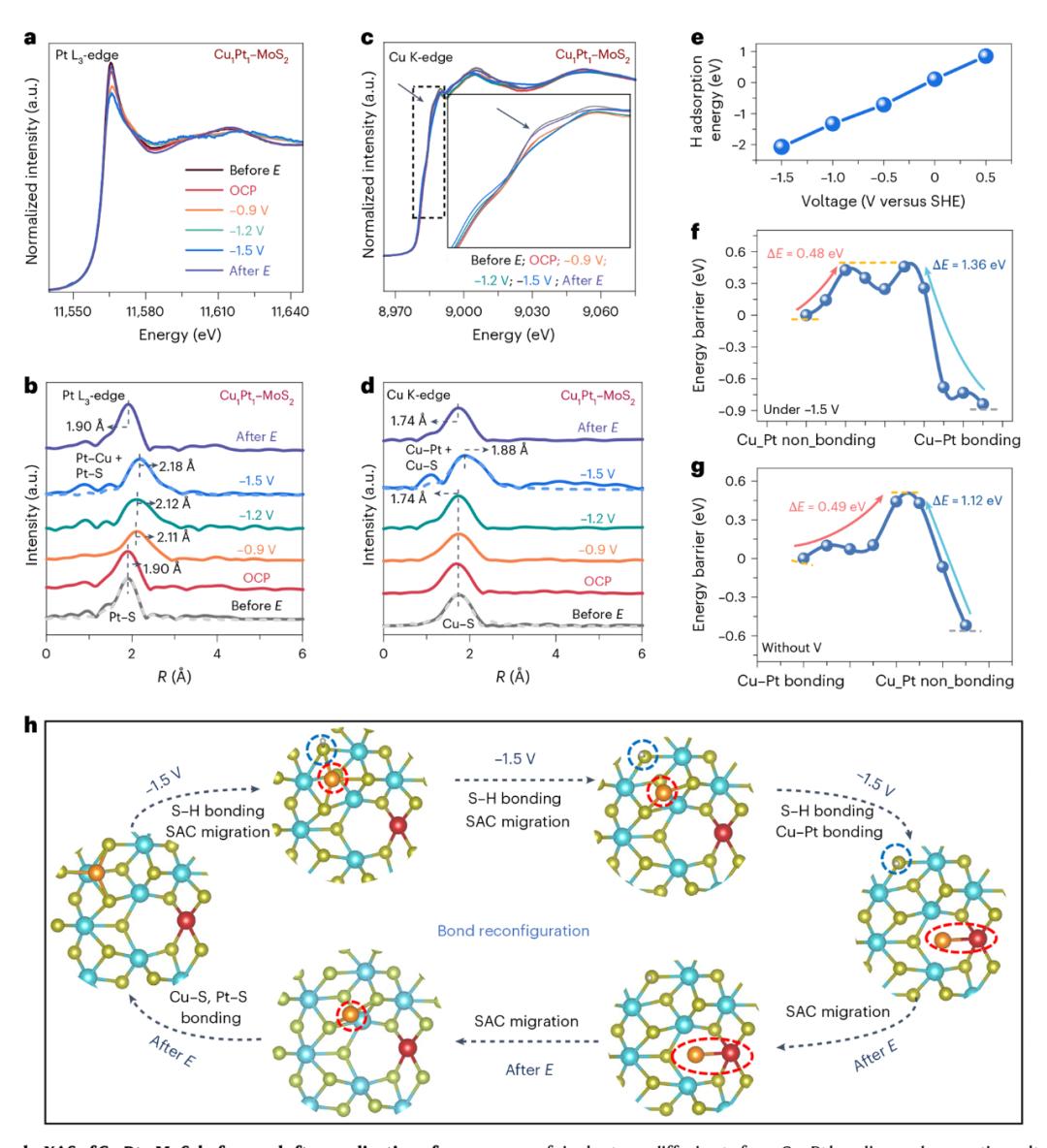
图 3. 电场诱导 Cu–Pt 按键过程的原点分析和机制建模。
图3显示了Cu围绕DAC的电场响应行为,重点是Cu–电压依赖性和可逆性在Pt成键过程中。原点 在负偏压压力下,XANES和EXAFS测量说明(–0.9 V至–1.5 V vs. Ag/AgCl)Pt在施加过程中L₃-在EXAFS峰顶,edge白线强度逐渐下降,–S的位置(~1.90 Å)向Pt–Cu键长度(~2.18 Å)转移,指示Pt–发生了Cu成键。Cu通道也表现出类似的行为,K-Cuuge谱峰位移和EXAFS信号变化共同表征–S键向Cu–转换Pt匹配状态。在电场拆除后,成键过程完全可逆,原子回归单原子分散态,表明DAC构型受到电场的精确调控。根据第一性原理计算,负电压诱导S原子质子化生成S–H键,从而减少金属–S键合强度,有助于金属原子的转移和重构。Cu迁移到Pt邻位的能垒约为0.48 eV,而且解键回归SAC状态的能垒为0.49 eV,确认构型转换具有高度可逆性和能量可控性。这张图清楚地描述了SAC在外场的影响下。–动态转换在DAC之间的完整路径,强调了电场对构型重构的“栅控效应”。
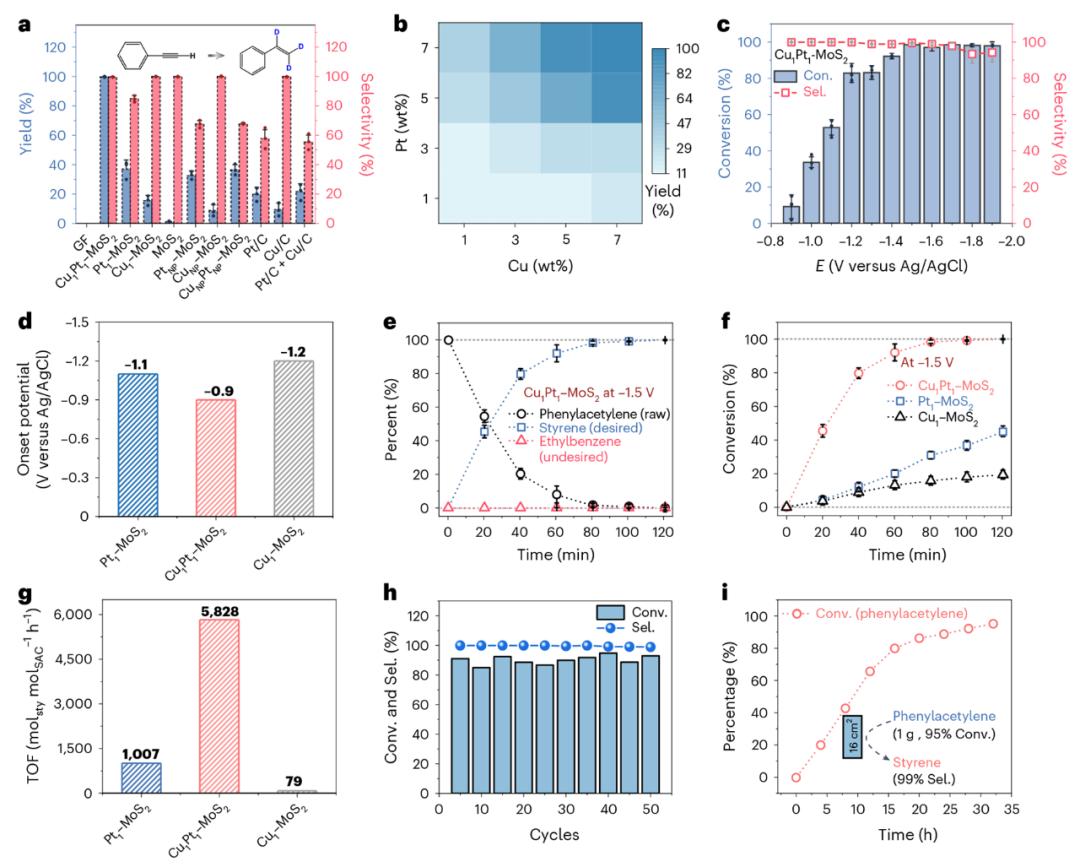
图 4. Cu–Pt DAC 性能表现为苯乙炔电催化邈代反应。
通过平台催化测试,图4验证了Cu–Pt 在电催化半氢化反应中,DAC表现出色。Cu1Pt1Pt1在苯乙炔的电化学反应中–MoS₂表现为近乎定量转化率(99%)和选择性,明显优于单一Cu或Pt SAC。相比之下,Pt SAC容易产生氢化,产生非目标产品;Cu 虽然SAC具有很高的选择性,但是整体活性较弱。DAC系统兼顾了两者的优势,–0.9 V至–1.7 V型电位窗表现出优异的稳定性和催化效率,起始电位显著移位,表明其反应热学门槛较低。80个DAC催化剂 反应可以在min内完成,单位点TOF高达5828 h⁻⊃1;,远远高于对照组。回收利用和克级反应系统中,仍然保持着高转化率和高选择性,显示出出色的操作稳定性和实际应用潜力。上述结果得到充分证实,Cu–在提高半氢化选择性和活性方面,Pt异核协同作用显著改善了反应路径和电子结构。
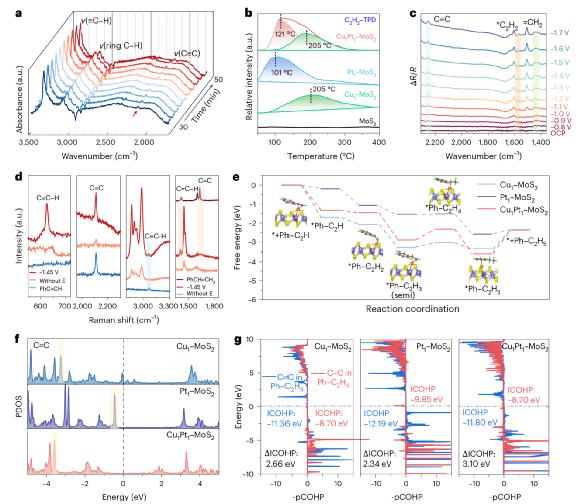
图 5. DAC 电子结构调节机制的催化反应路径。
图5系统阐述了DAC结构提高反应选择性的本质机制。通过DRIFTS和TPD试验,观察到苯乙炔具有较强的吸附能力和较高的DAC位点脱附温度,表明DAC提供了适当的吸附强度,有利于中间体的稳定存在而非过度反应。进一步分析了原点红外线和拉曼光谱。≡C与=C–H的动态振动模变化,揭示了DAC在电场下的活性过程和π按键功能调节特性。在DAC构型下,DFT能垒计算表明,从中间体丁二烯(C6H5)–进一步加氢为乙基苯所需的C2H3自由能障较高。( 0.56 eV),与Pt或Cu相比 SAC很难发生,从而提高了半氢化的选择性。PDOS和COHP分析指出,DAC位点诱导中间体电子态远离Fermi能级,C=C键与C键相比–C键持续时间较长,Bader电荷分析显示尾端碳位带负电增强,进一步抑制了过氢化的发生。上述结果系统揭示了DAC是如何通过调控电子结构的?π关键功能,轨道添加和反应自由能,实现对目标产品选择性的精确引导。
总结与展望
本研究基于1T缺陷工程调控。ʹ-MoS₂在此基础上,Cu和Pt单原子的超高负荷、稳定导向和空间邻居布局得以成功实现,异核双原子催化结构在反应条件下通过外加电场诱导其形成。(DAC)。原点 XAS Cu结合第一性原理运算分子动力模拟系统透露–Pt键的动态生成和可逆机制,明确其受S的影响–H键质子化状态调节。该可逆结构不仅赋予催化剂动态调节活性中心的能力,而且在电催化苯乙炔半氢化反应中表现出优异的活性、选择性和稳定性,明显优于传统的单原子或纳米粒子催化剂系统。此外,该系统具有较高的结构稳定性和实际工艺兼容性,已成功实现克级规模反应,呈现良好的放大前景。
展望未来,“电场可控构造重构”战略有望进一步扩展到更多的异种金属系统和其他二维媒体,推动电催化剂设计从“静态构造”向“动态可控构造”迈进。通过调节和反应外部条件,可以准确控制催化中心的电子状态、几何构造和反应路径,为构效关系研究、原子级机制分析和高性能电催化剂开发提供全新的范式。
免责声明:中国复合材料协会微信微信官方账号发布的文章仅用于复合材料理论知识和市场信息的交流与分享,不用于任何商业目的。如果任何个人或组织对文章版权或其内容的真实性和准确性有疑问,请尽快联系我们。我们会及时处理。
继续滚动阅读下一个轻触阅读原文。

学会向上滑动中国复合材料,看下一个。
原标题:“异核双原子催化剂动态调节新突破,电场控制下的“分分合合””
阅读原文
本文仅代表作者观点,版权归原创者所有,如需转载请在文中注明来源及作者名字。
免责声明:本文系转载编辑文章,仅作分享之用。如分享内容、图片侵犯到您的版权或非授权发布,请及时与我们联系进行审核处理或删除,您可以发送材料至邮箱:service@tojoy.com