
全球首款LAD-I基因疗法终获FDA批准,9名患儿重获新生,绝症迎来治疗新曙光





近日,美国食品药品监督管理局(FDA)正式批准Rocket Pharmaceuticals(NASDAQ: RCKT)研发的Kresladi(marnetegragene autotemcel)上市,该疗法适用于因ITGB2基因双等位基因变异引发的重症白细胞黏附缺陷症I型(LAD-I)儿科患者,且这些患者无法找到合适的人类白细胞抗原(HLA)匹配异体造血干细胞移植供体。这是全球范围内首个获批用于治疗LAD-I的基因疗法,也是近20年来LAD-I领域出现的首个全新治疗选择。
Kresladi的上市申请过程充满曲折。自2023年10月首次提交生物制品许可申请(BLA)起,该产品先后经历了审评延期、收到完整回应函(CRL)要求补充CMC信息、重新申报等多次挑战,最终在首次提交申请两年半后成功获批。FDA此次通过加速批准路径批准该产品,将中性粒细胞表面CD18和CD11a表达水平的提升作为替代终点。作为加速批准的条件,Rocket公司需开展上市后研究,进一步验证临床获益情况。
01 自体干细胞基因修饰技术,慢病毒载体递送ITGB2基因
白细胞黏附缺陷症I型(LAD-I)是一种由ITGB2基因突变导致的极罕见原发性免疫缺陷病。ITGB2基因负责编码CD18蛋白,该蛋白与CD11蛋白共同组成白细胞表面关键的黏附分子复合体——β2整合素。这一复合体是白细胞从血管内迁移至感染组织的必需功能单元。
在正常免疫应答过程中,当病原体侵入组织后,血液循环中的白细胞需先黏附于血管内皮细胞,再穿越血管壁进入组织,完成病原体清除。这一系列过程依赖白细胞表面β2整合素与血管内皮细胞表面黏附分子的相互作用。若ITGB2基因发生突变导致CD18蛋白功能缺失,β2整合素无法正常组装,白细胞便失去穿越血管壁的能力。
对于重症LAD-I患者,机体对细菌和真菌感染的防御机制会严重受损。患者从新生儿时期就开始反复出现严重的皮肤、呼吸道及深部组织感染,伤口愈合能力显著下降,感染难以控制且易复发。若不进行有效干预,多数患儿难以存活至儿童期。流行病学数据显示,LAD-I的发病率约为每百万活产婴儿1例,其中约三分之二为重症型。
目前,异体造血干细胞移植(骨髓移植)是部分重症LAD-I患者的治疗选择,但该方案存在两大限制:一是需找到HLA匹配的同胞供体,临床实践中成功率较低;二是移植过程伴随清髓性化疗、移植物抗宿主病(GVHD)等严重并发症风险,治疗相关死亡率较高。对于无合适供体的患者,长期以来缺乏有效治疗手段。
Kresladi属于体外基因修饰自体造血干细胞疗法,其技术路线可概括为:采集患者自身的造血干细胞,在体外利用慢病毒载体将正常功能的ITGB2基因拷贝导入细胞内,使干细胞具备表达功能性CD18蛋白的能力;通过清髓性化疗预处理清除患者体内原有缺陷造血干细胞后,将基因修饰后的细胞回输至患者体内;回输的干细胞在骨髓中定植并持续分化为功能正常的白细胞,从根源上修复免疫缺陷。
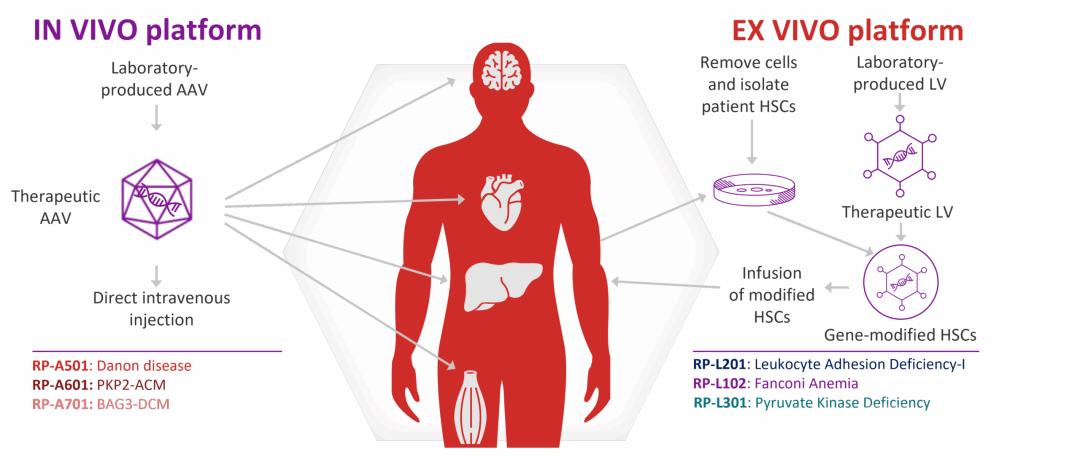
该疗法的核心研发工作由西班牙CIEMAT(国家能源、环境与技术研究中心)和Fundación Jiménez Díaz医院研究所联合开展,并与英国伦敦大学学院(UCL)合作完成慢病毒载体开发。2019年,Rocket Pharmaceuticals从上述机构获得该疗法的全球授权,并在美国推进临床开发。UCLA的Donald Kohn博士作为该领域资深研究者,主导了北美地区的临床试验。
由于使用患者自体细胞,该疗法理论上规避了异体移植中常见的免疫排斥和移植物抗宿主病风险。
02 9例患者生存率100%,感染率显著降低
Kresladi的上市申请主要基于编号为NCT03812263的全球多中心I/II期临床试验。该研究为开放标签、单臂设计,共纳入9例重症LAD-I患儿,年龄在5个月至9岁之间,均为ITGB2基因双等位基因变异导致的重症患者,且无合适HLA匹配同胞供体。患者分别在UCLA(6例)、英国伦敦(2例)和西班牙(1例)三个中心接受治疗。
研究主要终点为输注后12个月的生存率,以及中性粒细胞表面CD18和CD11a的表达水平;次要终点包括严重感染发生率、皮肤病变改善情况、伤口愈合能力等。
KRESLADI的数据显示LAD-I的生存率为100%,且疗效显著
研究结果表明,所有9例患者在输注后12个月及整个随访期(18至42个月)均存活,生存率达100%。治疗期间无患者需要进行异体造血干细胞移植,未出现移植物衰竭或免疫排斥事件。
治疗后,患者中性粒细胞表面CD18和CD11a的表达水平显著提升。这一指标作为功能性β2整合素恢复的生物标志物,被FDA采纳为替代终点,为加速批准提供了支持。
在临床结局方面,与治疗前基线相比,患者的严重感染发生率显著下降,皮肤病变有所改善,伤口愈合能力得到恢复。部分患者在接受治疗后逐渐脱离了长期的抗生素预防治疗。
安全性方面,Kresladi总体耐受性良好。研究期间未报告与治疗相关的严重不良事件。患者接受的清髓性预处理化疗相关不良反应可控,未出现移植物抗宿主病等异体移植特有的并发症。相较于传统异体造血干细胞移植方案,该疗法的短期和长期毒性特征更具优势。
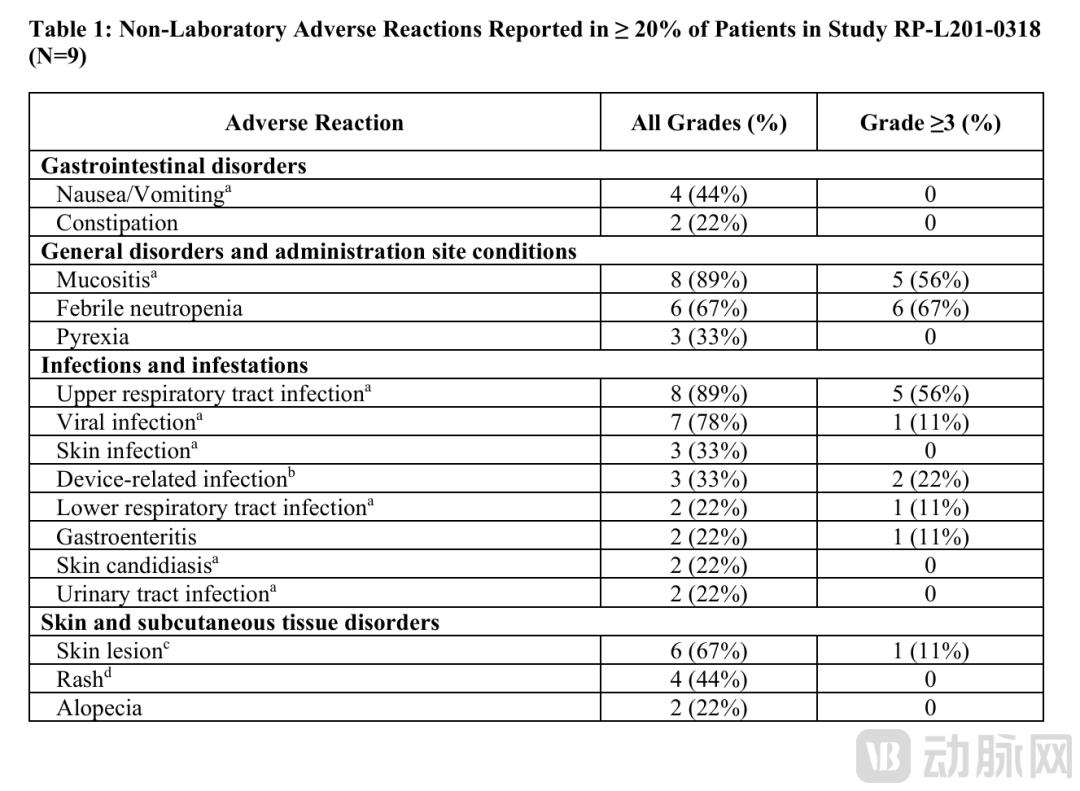
常见不良反应报告
03 两次审评延迟,加速批准附带上市后研究要求
尽管临床数据明确,Kresladi的上市审评过程并不顺利。
2023年10月,Rocket向FDA提交BLA申请,获得优先审评资格,PDUFA日期定为2024年3月31日。此前,Kresladi已获得再生医学先进疗法(RMAT)、罕见儿科疾病、快速通道等多项资格认定。
2024年2月,FDA因审评人员变更需要额外时间审阅CMC信息,将PDUFA日期延至2024年6月30日。
2024年6月28日,FDA向Rocket发出完整回应函(CRL),要求补充“有限的额外CMC信息”。这是FDA第二次就CMC问题提出补充要求,消息公布后Rocket股价单日跌幅超11%。但Rocket在后续与FDA的沟通中确认,所需补充信息范围有限,且不涉及临床疗效或安全性数据的重新评估。
经过一年多的CMC数据补充和整改,Rocket于2025年10月14日重新提交BLA申请,FDA将新的PDUFA日期定为2026年3月28日。
2026年3月26日,FDA正式批准Kresladi上市,比预定PDUFA日期提前两天。
FDA此次通过加速批准路径批准该产品,将中性粒细胞表面CD18和CD11a表达水平作为替代终点。这一生物标志物与LAD-I患者免疫功能的恢复密切相关,被认为可合理预测临床获益。该替代终点的选择与RMAT认定中监管机构与申办方的沟通结果一致。
作为加速批准的条件,Rocket需开展上市后研究,继续收集长期随访数据,进一步验证Kresladi在真实世界中的临床获益。若上市后研究数据无法支持临床获益,FDA保留撤销批准的权利。
此外,FDA同时授予Rocket罕见儿科疾病优先审评凭证(Priority Review Voucher,PRV)。该凭证可转让或出售,目前市场估值约2亿美元,可为Rocket提供一笔非稀释性资金支持。
04 极罕见病基因疗法探索监管新路径
Kresladi的获批对体外基因修饰细胞药物领域具有多重意义。
首先,该产品凭借9例患者的临床数据获得FDA批准,进一步验证了在极端罕见病领域,监管机构对“充分满足未满足医疗需求”的考量权重正在提升。在临床证据足够清晰、替代终点选择合理的前提下,小型单臂试验的数据可支撑上市批准。这一信号对其他极罕见遗传免疫缺陷病的基因治疗开发具有参考价值。
其次,Kresladi的审评历程凸显了CMC(化学、制造与控制)在细胞基因疗法开发中的重要性。该产品的临床疗效数据始终未受质疑,但CMC问题导致上市申请延后两年有余。对于自体造血干细胞基因修饰疗法,每例患者的起始细胞存在个体差异,基因修饰过程涉及慢病毒载体的生产和细胞转导,对工艺稳定性、批次一致性、质量控制体系提出极高要求。基于此,对于生物科技企业而言,CMC能力已从“可晚些考虑的配套环节”转变为“与临床开发同步推进的关键基础”。
关于Kresladi的商业化前景,重症LAD-I全球每年新发病例仅数十例,患者群体规模极小,该产品的市场体量有限。据William Blair分析师估计,Kresladi的年销售峰值约为2.94亿美元。Rocket需通过定价策略、医保覆盖和专业治疗中心网络的建立,实现可持续的商业运营。同时,加速批准附带的上市后研究要求,也意味着企业需要持续投入资源以维持上市资格。
本文来自微信公众号“动脉网”(ID:vcbeat),作者:宁晨,36氪经授权发布。
本文仅代表作者观点,版权归原创者所有,如需转载请在文中注明来源及作者名字。
免责声明:本文系转载编辑文章,仅作分享之用。如分享内容、图片侵犯到您的版权或非授权发布,请及时与我们联系进行审核处理或删除,您可以发送材料至邮箱:service@tojoy.com