
清华团队研究:揭秘锂电池电解液中锂键特性与规律






【研究背景】
理解锂与配位组分间的相互作用,对阐明锂电池内部工作机制(如离子传输动力学、界面反应机制)及指导新一代锂电池研发至关重要。锂键作为描述锂和配体之间非共价相互作用的概念,虽在2010年代引入电池领域研究,且在缓解锂硫电池正极“穿梭效应”、调控聚合物电解质溶剂化结构等方面取得成功,但因真实电池体系复杂,工作电池中锂键特性未充分探索,其受电解液组成调控的规律及与常规离子键的根本差异尚不明确,这制约了锂键理论的发展和应用。
【工作简介】
近日,清华大学化学工程系张强、陈翔等通过分子动力学(MD)模拟、密度泛函理论(DFT)计算与机器学习方法,系统探究锂电池电解液中的锂键特性并建立普适性规律。研究构建不同阴离子浓度、含不同有机溶剂和阴离子的多种有机电解液体系,验证锂键在电解液中的普遍存在性,揭示高盐浓度(HC)可显著提升锂键占比(部分体系超40%)与平均寿命(部分体系超1.0ps)。通过核磁共振(NMR)谱学计算,明确配位数(CN)为4是区分锂键与离子键的通用分界点:CN在1 - 4范围时,体系由锂键主导;CN增加到5 - 6时,转变为离子键主导。对锂周围电子环境分析,发现上述变化对应锂离子周围电子局域化区域因配位偏离球形对称,揭示两者在电子环境与成键特性上的核心差异。最后构建集成机器学习模型,以组分加权平均最高占据分子轨道 – 最低未占分子轨道(HOMO–LUMO)能级差为关键描述符,实现对<7>LiNMR化学位移的高精度预测(测试集决定系数为0.93),证实阴离子对锂键特性的影响显著大于溶剂,为先进锂电池电解质的理性设计提供理论支撑与预测工具。该成果发表在化学领域顶级期刊Angew. Chem.上,清华大学硕士研究生高岩斌和本科生李蔚林为本文共同第一作者,通讯作者为陈翔副研究员和张强教授。
【内容表述】
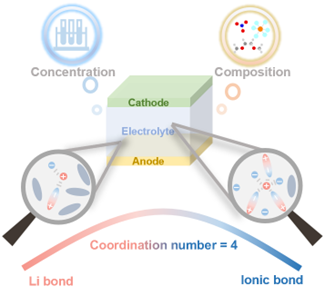
Figure1.锂电池电解液中的锂键特性及区分其与离子键的普适性规律。
锂键是类似氢键的次级相互作用,但锂的金属性使锂键具较强离子键特性,行为比氢键复杂。如一个锂原子最多可配位六个原子,而一个氢原子通常只涉及一个氢键和一个共价键。因此,厘清锂键特性及其与常规锂离子键的区别很重要。受NMR光谱表征氢键化学环境的启发,<7>LiNMR被用作探索锂键化学的主要手段。此前研究表明,锂键形成会使锂的化学位移向低场移动。在特定电解液体系(DME和FSI⁻)中,随CN增加,<7>LiNMR化学位移呈“火山型”曲线,对应体系从锂键主导向离子键主导的转变,为本研究奠定基础。
3.1电解液中锂键的动态演化过程分析
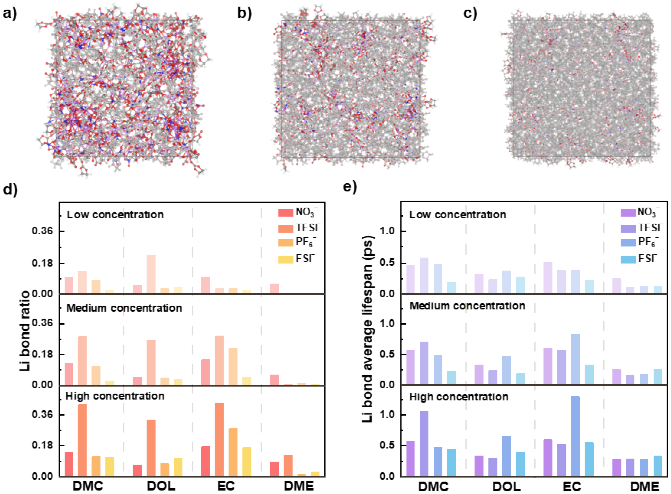
Figure2.低、中、高盐浓度电解液的构筑及其中锂键的性质分析。
为证实锂键广泛存在并探究其形成规律,研究构建包含4种常见溶剂(DME、DOL、EC和DMC)和4种常见阴离子(NO₃⁻、PF₆⁻、TFSI⁻和FSI⁻)的48种电解液模型,涵盖低、中、高(1.05,2.01和4.02M)三种盐浓度。通过分子动力学模拟追踪统计锂离子在不同配位环境下的动态演变过程,验证锂键在电解液体系中的普遍存在性,发现盐浓度对锂键特性影响显著:随浓度从低到高增加,体系中锂键占比和平均寿命普遍提升。部分体系锂键占比从约3%增至超40%,平均寿命从约0.3ps延长至超1.0ps。对溶剂化结构分析表明,高盐浓度下阴离子更易进入锂离子第一溶剂化层,形成带负电的团簇结构,阻碍外部离子、分子渗透,增强锂键稳定性。
3.2锂键与锂离子键的区别和对比分析
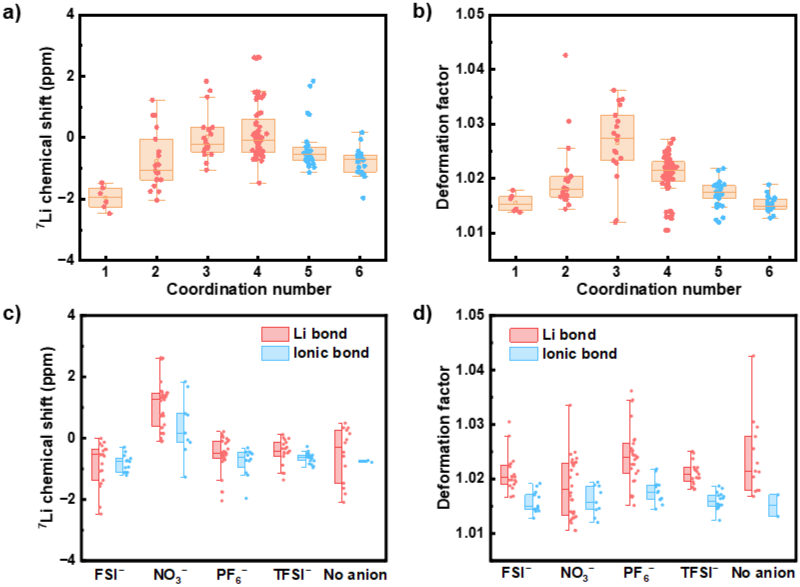
Figure3.锂键和锂离子键的对比分析。
DFT计算揭示锂键与离子键的本质区别。研究发现<7>LiNMR化学位移呈普适的“火山型”变化趋势:锂的化学位移在CN = 4前后先增大后减小,区分了锂键(CN = 1、2、3或4)与离子键(CN = 5或6)主导的两种相互作用区域。深入分析发现,锂离子周围电子局域化区域对称性的变化是化学位移非单调行为的关键因素。此外,研究发现阴离子种类(如NO₃⁻)对<7>LiNMR化学位移的影响远大于常见溶剂分子,且这种阴离子的特异性影响无法单纯由电子局域化区域对称性的变化解释。
3.3数据驱动的锂键性质探究
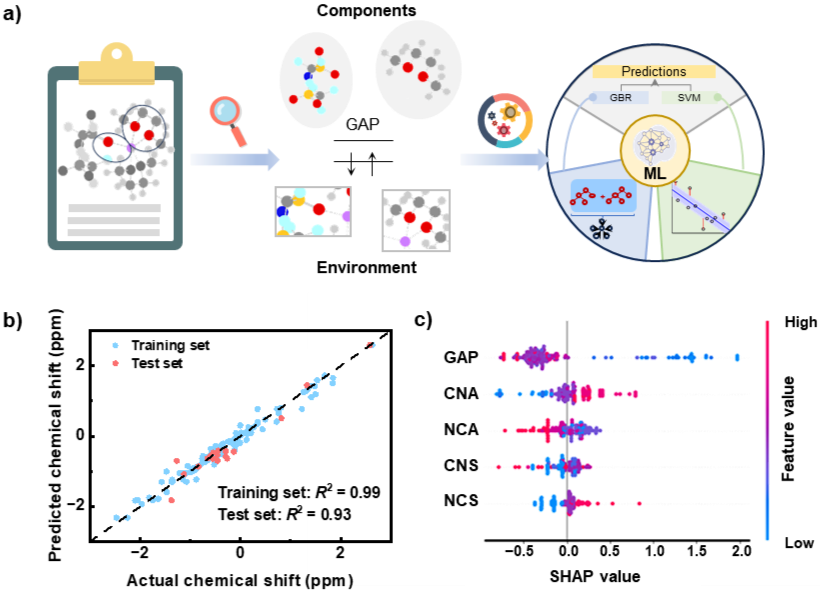
Figure4.基于DFT计算结果的机器学习模型的构建与分析。
研究者开发可解释的机器学习模型,探究电解液组分调控锂键特性的深层原因,发现溶剂化结构中阴离子(NCA)和溶剂数目(NCS),阴离子(CNA)和溶剂各自的配位数(CNS),以及组分加权平均HOMO–LUMO能级差(GAP)是重要特征,建立能高精度预测<7>LiNMR化学位移的集成学习模型,并通过Shapley值(SHAP)分析方法揭示各输入特征对化学位移的贡献度,发现配位组分的加权平均HOMO–LUMO能级差是影响化学位移的最关键描述符,突显阴离子在调控锂离子局域电子环境中的重要地位。
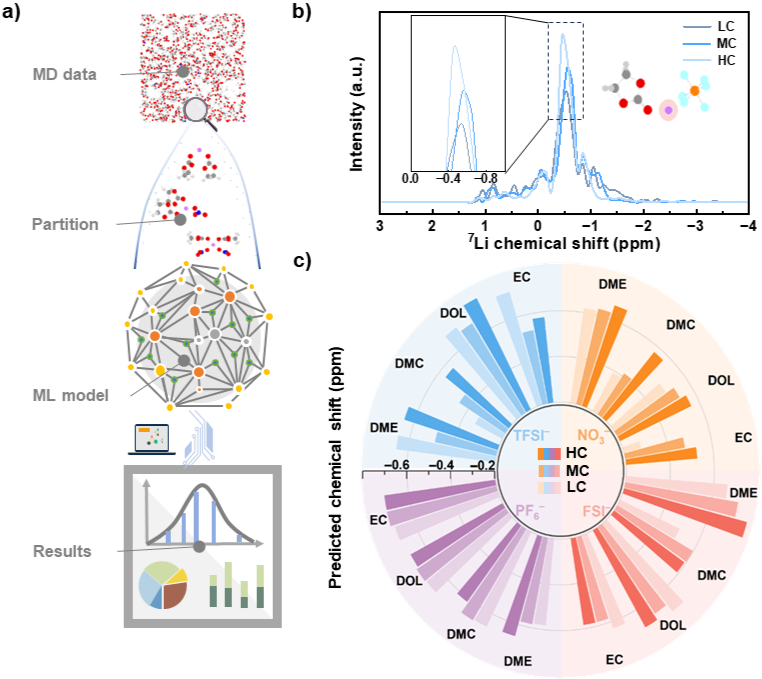
Figure5.基于MD模拟结果的机器学习模型预测。
最后,研究者将MD模拟与机器学习模型相结合,成功预测48个电解液体系内<7>LiNMR化学位移的分布和平均化学位移,验证盐浓度对化学位移的影响趋势,与先前研究的计算和实验趋势吻合。这表明该模型不仅能定量关联原子级溶剂化结构与实验光谱数据,还可用于高通量筛选具有定制化溶剂化结构与高离子扩散率的先进电解液,为下一代高性能锂电池材料的理性设计提供高效计算工具与理论指导。
【结论】
本工作通过融合多尺度理论计算与可解释的机器学习,首次系统性建立锂电池电解液中锂键特性的普适性原理。研究明确锂键在多种常规电解液体系中的普遍存在性,高盐浓度环境会增强阴离子与Li⁺的配位,提升锂键的占比与平均寿命。确立区分锂键与离子键的判据:CN为1 - 4时锂键主导,CN为5 - 6时离子键主导。解析组分影响,发现阴离子对<7>LiNMR化学位移的影响远大于溶剂,构建高精度机器学习预测模型,发现电解液组分的加权平均HOMO–LUMO能隙是预测化学位移的最关键描述符,为电解液的理性设计提供高效计算工具和理论指导。本研究深化对锂键的科学认知,所建立的普适性原理和预测模型,为高性能锂电池先进电解质材料的设计与开发奠定理论基础。
本文仅代表作者观点,版权归原创者所有,如需转载请在文中注明来源及作者名字。
免责声明:本文系转载编辑文章,仅作分享之用。如分享内容、图片侵犯到您的版权或非授权发布,请及时与我们联系进行审核处理或删除,您可以发送材料至邮箱:service@tojoy.com